
A metabolomics-based method for the analysis of wheat grain
Hayley Abbiss 1,2, Michael Francki 3,4, Grantley Stainer 3, Robert French 3, Joel P.A. Gummer 1, Scott J. Campbell 5, John H. Moncur 5 and Robert D. Trengove 1
1 Research and Innovation, Murdoch University, Perth, WA, Australia, 2 Centre for Digital Agriculture, Curtin University, Perth, WA, Australia, 4 Department of Primary Industries and Regional Development, Perth, WA, Australia, 5 State Agricultural Biotechnology Centre, Murdoch University, Perth, WA, Australia and 5 SpectralWorks Ltd, United Kingdom,
First Published: 40th BMSS Annual Meeting 2019 (Manchester, UK).
Introduction
For mass spectrometry (MS)-based metabolomics it is recommended to include the analysis of reference samples (preferably pooled study samples), as an added quality control (QC) measure at regular intervals throughout an analytical sequence. The reference can be used to measure changes in signal and a correction algorithm can later be applied to sample metabolite measurements, to monitor introduced technical variation effecting metabolite concentration. There are few single software platforms for data processing, signal-correction and analysis/interpretation which are vendor neutral and support GC- and LC-MS data, and fewer implemented through a GUI. Here we present this software capability and compare correction methods using a sample set of wheat grain analysed by LC-Q-ToF-MS.
Methods
Ten varieties of wheat were field-grown in triplicate in Merredin, Western Australia. Mature grain was harvested and processed for metabolite analysis. Briefly, metabolites from 200 mg of freeze dried, finely ground grain were extracted using acetonitrile (1.5 mL) containing four internal standards (IS; 2-aminoanthracene, miconazole, 13C6-sorbitol, d6-transcinnamic acid). An aliquot (50 µL) of the extract was diluted to 5% acetonitrile (950 µL) with water containing the injection standard, leucine-enkephalin. Validation samples (8 IS concentrations, 3 technical replicates of each) were prepared from grain pooled from each study sample (n=30). QC samples (prepared from both pooled grain and pooled extract) were spaced evenly throughout the analytical sequence and validation samples were analysed in blocks at the beginning, middle, and end of the sequence. Validation blocks were used to assess data quality (linearity, reproducibility) after QC correction. A Waters Acquity LC system equipped with a quaternary solvent manager and Waters HSST3 column were used, and coupled to a Sciex 5600 Triple ToF acquiring in both positive and negative ToF-MS modes over the m/z range 50 -1,300.
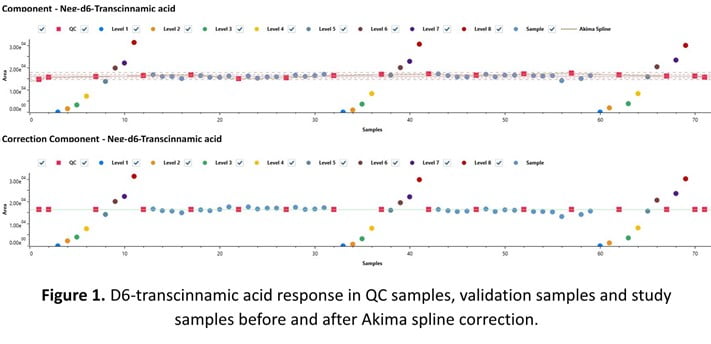
Results
A QC correction tool recently implemented into the vendor neutral data processing software, AnalyzerPro®, is presented. AnalyzerPro offers two spline types for signal correction. The first is the Akima spline (shown in Figure 1) and second a smoothing spline with the smoothing level set by the user (0 = not smoothed, 1 = highest level of smoothing). These methods are demonstrated here (Figures 1-3).


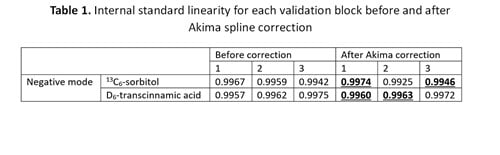
For negative mode validation block data, the Akima spline was optimal with an average IS RSD (%) of 2.28% (Figure 3). The linearity of internal standards was also improved with four of six R2 values improved after Akima spline correction (Table 1).
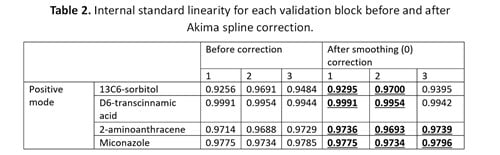
For positive mode validation block data, the best results were observed for no correction (Figure 3). However, a smoothing spline correction set to 0 gave the optimal linearity of internal standards with ten of twelve R2 values equal to or greater than the uncorrected data (Table 2).
Conclusion
In general, linearity and reproducibility of quality assessment samples was improved by correcting data. Including samples (containing standards) to validate not only the MS analysis but the data processing pipeline is necessary for the assessment of data quality.
Acknowledgements
The Premier’s Fellowship Program for Agriculture and Food (Department of Jobs, Tourism, Science and Innovation, Western Australia), Murdoch University and the Premier’s Fellow, Professor Simon Cook, are acknowledged for supporting this work. The National Collaborative Research Infrastructure Strategy-funded Bioplatforms Australia (BPA) is also acknowledged.