
Scott J Harrison1, J Richard Dickinson1 and Scott J Campbell2
1University of Cardiff, Wales, UK; 2SpectralWorks Ltd, The Heath Business & Technical Park, Runcorn, UK
First Published: BMSS 2005
Introduction
With the advent of both the human genome project and the science of genomics there has been great advances in the level of knowledge about genes and gene function. Several species of plants, animals and microorganisms have now had their genomes fully mapped. However the fact that the genome is mapped does not imply that we know the function of many of the genes. Yeast (Saccharomyces cerevisiae) is a model single cell organism which is used for scientific studies. The entire genome of Saccharomyces cerevisiae has been published in a special edition of Nature. Transcriptomics allows us to measure which genes within an organism are being transcribed and can be used to infer gene function. There are many more transcripts than genes so the story becomes more complex.
Most cellular processes are carried out by multi-protein complexes. The identification and analysis of their components provides insight into how the expressed proteins (proteome) are organized into functional units.
When we examine the reported small molecules that have been reported as primary metabolites, we find a vast simplification of the number of possible chemical species. In fact the number chemical species reported with the literature is in the order of 3000. However both the physical and chemical properties of the molecules vary greatly, therefore, it is not possible to analyse all the molecules of the metabolome in a single method. Currently the attempts to study the small molecule compliment (metabolome) divides into two groups. First, one approach is to look for differences in the patterns of unidentified analytes using statistical methods such as principle component analysis (PCA), this is also know as Metabonomics and is not related to the underlying biochemistry. The second type of analysis is based on the monitoring of all the possible compounds and relating them back to the underlying biochemistry, this is known as Metabolomics.
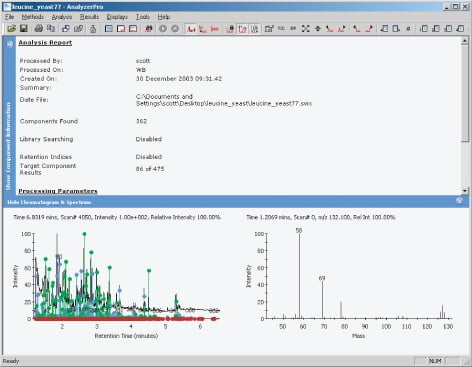
Figure 1.
Metabolomics in its purist form would monitor the entire small molecule compliment. However, this has proved to be more complex than it sounds, as each extraction and analysis technique is selective and suited for particular compound classes. For these reasons, most people who are currently practicing metabolomics are doing so in a form that is more similar to that used in classical analytical chemistry. The techniques being used are those that are commonly known as target compound analysis, in which a list of target compounds are analysed for within a mixture.
The compounds in the list are run as standards, to monitor both analytical and chromatographic efficiencies. If the target compounds are carefully chosen, it is possible to monitor pathways and the underlying biochemistry. The data analysis with the target compound analysis is relatively simple using the software available from the instrument vendors or other third-party software. This approach by definition incurs the loss of data from the compounds that are not being analysed within the target compound list. There are only two commercially available software packages that analyse data files to extract the data for the entire components within the mixture. Both packages rely on similar mathematical algorithms to extract the peak information from the data files, this approach was pioneered by NIST. The principles used are known as deconvolution.
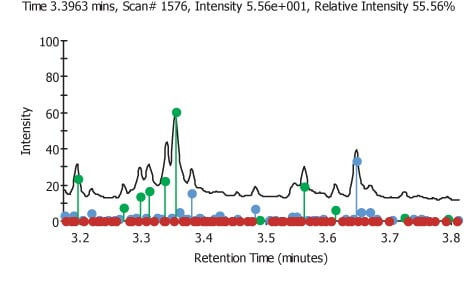
Figure 2.
Materials and Methods
Various yeast strains were grown under identical growth conditions. The strains were single gene knock-outs derived from the same parent strain. They were grown in the same media type to the same cell densities. To analyse the whole cells, the cells cultures were then centrifuged and the media was pipetted from the cell pellet. The pellet was then resuspended in ice cold distilled water and centrifuged. The water was pipetted from the pellet and the cells were washed two further times. The cell pellets were then frozen at -80°C prior to aliquoting the samples for analysis. The cell pellets were thawed and 20 µl of the pellet was aliquoted into a glass vial held in a 96-well plate and sealed with a press fit mat. The plates were then frozen at -80°C and were freeze dried. Once dried the plates were stored in a vacuum chamber containing silica gel desiccant prior to derivatisation. The plate was transferred to a dry box containing an inert dry nitrogen atmosphere, where they were derivatised with a base catalysed TMS reagent. Once derivatised the sealing mat was replaced whilst still in the dry box the plate was then removed from the dry box and placed on the autosampler being analysed immediately. The derivatised samples were analyzed on a Thermo Electron Tempus Time of Flight mass spectrometer, coupled to a Thermo Electron Trace2000 Gas Chromatograph and a CTC Combi PAL liquid autosampler, under the following conditions.
The data files were examined using AnalyzerPro from SpectralWorks (Figure 1), to find all the peaks present within the total ion current (TIC). The retention time and spectra under these peaks were used to create a processing method, to allow the identified peaks to be quantified. The quantification was carried out in a relative sense rather than an absolute. Each individual spectrum was searched against the National Institute of Standards (NIST) database of spectra, to aid identification. Each file gave an area for the 51 peaks or components found under the TIC, all the areas for each peak were placed in a spreadsheet and were analysed. The areas for each component were all normalised by dividing the area by the average area for that component.
The normalised areas for each file were plotted on a bar chart against the file number. Error bars equivalent to three times the standard deviation from the average value of the area for a given component. Only components that were over three standard deviations were chosen as being significantly up regulated. One potential weakness in this approach can be seen by examination of the component graphs. Any component that is down regulated when compared to the average will not appear to be significant as many of the error bars minimum value is negative and if the component is not detected then the value of the area that is measured will be zero, meaning the relative area will be zero but of course, this is still within the limits set by the error bars. Of the 51 components that were examined by this method 23 were found to have at least one or more files that were significantly different. In Figure 2 we can see the areas on the TIC were found target components are shown in green, new components are shown in blue and non found target components are shown in red
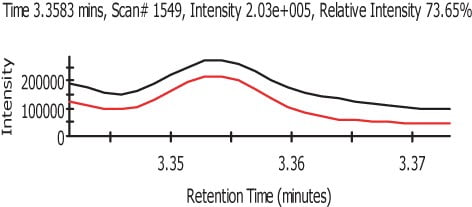
Figure 3.
The procedure is designed to yield a very broad range of metabolites while not specifically targeting any particular analyte or class of analytes. Dolan stated that developing a stability-indicating assay requires consideration of three aspects of the method: obtaining a representative sample, choosing the separation technique and selecting the detector. To maximize the number of compounds that could be analysed, a derivatisation process was used; the procedure was designed to react with any labile hydrogens (such as those on alcohol, amines and acids) replacing the hydrogen with a silane group (Si(CH)3). The derivatisation procedure creates multiple products due to the multiple sites of silanization. The reaction also does not necessarily go to completion for all analytes, nor are the extent of side reactions fully known.
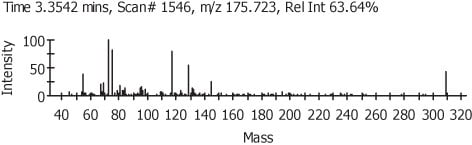
Figure 4.
Results and Discussion
We can see from Figure 2 that the chromatograms are very complex, the ability to reliably and rapidly analyze the data files by use of AnalyzerPro made this comparison of the components possible. The data set of 100 files could be analyzed within an hour and the use of the MatrixAnalyzer plug-in enables the files to be conveniently and rapidly compared.
Once the files had been processed the results were placed into a spreadsheet. The components were then compared against the average, differences were deemed to be significant if the value varied more than three standard deviations from the mean. The approach however seems to only be able to find up regulated components, as in most cases the value for the minus three standard deviation line is negative. A negative value for either area or height is of course impossible and therefore means we can not monitor for down regulations on this dataset. A possible way to improve this situation is to increase the number of replicates and therefore reduce the standard deviations of each component’s average area.
In Figure 3 we can see the representation of the TIC in black and the refined TIC from AnalyzerPro in red. Figure 4 shows the automatically refined spectra and Figure 5 show the identification of the spectra when search against the NIST02 spectral library database.
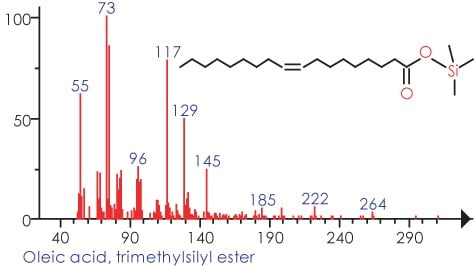
Figure 5.
Conclusions
As stated previously it appears that a gene deletion need not necessarily lead to a discernible phenotype and from the results we obtained, it appears that many gene deletions are also biochemically silent. The biochemical silence of many of the gene deletions is probably a result of the fact that many of the enzymic systems have duplications. We have shown that it is possible to measure up regulation of biochemical intermediate; however, with these experiments it has not been possible to monitor down regulation of metabolites. It is important that a method is found to find analytes that are down regulated; to do this the standard deviation needs to be reduced, the number of replicates needs to be increased. Although not mentioned previously LC-MS was tried on the yeast samples but the methods need to be examined in greater detail. The current methodologies have shown promise but have not conclusively given any clear answers. One of the problems is the wide standard deviations of the data, to reduce the standard deviation it is recommended that the strains be re-grown in larger quantities and that these samples are re-analysed several times. The data mining relied on target compound analysis, which means that only large peaks are analysed and many less abundant components may be missed.